1. Introduction
RNApedia is a database of protein–RNA complexes
RNApedia is a publicly available database designed to store, organize, and analyze three-dimensional (3D) structures of protein–RNA complexes. The platform provides a unified environment for accessing structural, sequence-based, and interaction-level information, enabling systematic studies of protein–RNA recognition and binding mechanisms.
RNApedia is available at:
https://bioinfo.dcc.ufmg.br/rnapedia3/
What is RNApedia?
RNApedia is a curated database dedicated to protein–RNA interactions, focusing on experimentally resolved complexes deposited in the Protein Data Bank (PDB)[1]. Unlike general structural repositories, RNApedia organizes data at the protein–RNA pair level, allowing precise characterization of interactions between individual protein chains and RNA molecules.
The primary goal of RNApedia is to provide a standardized and interaction-centered resource that supports both biological interpretation and computational modeling of protein–RNA complexes. The database was designed to facilitate large-scale analyses, comparative studies across RNA classes, and the development of predictive models, including machine learning and artificial intelligence approaches.
1.1 Overview
Protein–RNA interactions play a central role in essential cellular processes such as transcriptional regulation, RNA processing, translation, ribosome assembly, and post-transcriptional control. Despite the availability of raw structural data in the PDB, protein–RNA complexes remain challenging to analyze systematically due to structural heterogeneity, variable annotation standards, and the lack of interaction-focused organization[2].
RNApedia addresses these challenges by extracting, curating, and enriching protein–RNA complexes with uniform annotations, RNA classification, interaction metrics, and structural descriptors. Each entry in the database represents a well-defined protein–RNA interaction pair, enabling consistent comparisons across complexes.
The platform integrates an intuitive web interface with advanced search and exploration tools, allowing users to query complexes by PDB identifiers, RNA type, experimental resolution, interaction features, and sequence-derived properties. All data are freely available, promoting reproducibility and broad reuse within the scientific community.
Key Highlights
-
Curated protein–RNA dataset
Systematically organized protein–RNA complexes derived from experimentally determined structures. -
Interaction-centered organization
Data structured at the protein–RNA pair level, enabling precise analysis of molecular interfaces. -
RNA classification support
Integration of RNA type information to facilitate functional and comparative studies. -
Advanced search and exploration
Flexible querying and filtering across structural, sequence, and interaction attributes. -
Open-access platform
Freely available data and downloads, supporting transparent and reproducible research.
1.2 Database content and scope
RNApedia encompasses protein–RNA complexes resolved by experimental methods such as X-ray crystallography, cryo-electron microscopy, and NMR spectroscopy. The database includes a diverse range of RNA classes, including ribosomal RNAs, transfer RNAs, messenger RNAs, and non-coding RNAs, reflecting the broad functional landscape of protein–RNA interactions.
For each complex, RNApedia provides:
- Structural metadata (resolution, experimental method)
- Protein and RNA sequence information
- RNA classification and annotation
- Interaction-related features and contact metrics
- Direct access to structure visualization and analysis tools
1.3 Intended applications
RNApedia was designed to support a wide range of applications, including:
- Structural and functional analysis of protein–RNA interfaces
- Comparative studies across RNA classes
- Identification of conserved interaction patterns
- Benchmarking and training of machine learning models
- Development of predictive tools for protein–RNA recognition
2. How to use RNApedia
2.1 Home page
RNApedia provides an integrated web interface that enables access, exploration, and analysis of protein–RNA complexes in a systematic and user-friendly manner. The homepage of the platform (Figure 1) was designed to centralize the main functionalities, allowing users to quickly initiate data exploration or targeted analyses.
As shown in Figure 1, the top navigation bar organizes the core modules of RNApedia, including institutional information (About), technical documentation (Documentation), data download options (Download), data exploration (Explore), advanced querying (Advanced Search), and a dedicated tool for protein–RNA contact analysis (Contact Analysis Tool). This modular organization facilitates a clear transition between data retrieval, exploration, and analysis workflows.
Also on the homepage (Figure 1), the quick search field enables direct retrieval of specific complexes using a PDB identifier, an internal RNApedia identifier, or chain-specific notation. This feature is particularly useful for users who already have a predefined complex of interest.
The homepage further provides shortcut buttons to the Explore and Documentation sections (Figure 1), offering immediate access to data browsing and methodological descriptions of the database. These shortcuts reduce the number of steps required to begin using the platform, especially for new users.
In addition to navigation elements, the homepage displays a schematic representation of a protein–RNA complex, emphasizing the focus of RNApedia on three-dimensional structures and molecular interaction interfaces. Information on how to cite the database is also provided, ensuring appropriate acknowledgment of RNApedia in scientific publications.

2.2 Data download in RNApedia
RNApedia provides open access to all curated datasets through a dedicated Download page, enabling users to retrieve structural, sequence-based, and annotation data for offline analysis and large-scale computational studies. The Download interface (Figure 2) was designed to clearly present the available datasets, their formats, and file sizes, facilitating data reuse and reproducibility.
As shown in Figure 2, each dataset entry is organized into three main columns: file type, file size, and download action. This layout allows users to quickly assess the format and storage requirements of each dataset before initiating a download.
The RNApedia datasets include a comprehensive tabular file listing all curated protein–RNA complexes, complete datasets containing structural files, sequences, and annotations, as well as specialized subsets focused on binding affinity data, RNA-binding structural motifs, and RNA modifications (Figure 2). In addition, RNApedia provides separate FASTA files for protein and RNA sequences, enabling straightforward integration with sequence-based analyses and external bioinformatics pipelines.
All datasets are distributed in standard formats, such as TSV, ZIP, and FASTA, ensuring compatibility with commonly used bioinformatics tools and workflows. This modular organization allows users to download only the datasets relevant to their specific research questions, ranging from exploratory analyses to large-scale modeling and machine learning applications.

2.3 Advanced Search on RNApedia
Advanced Search in RNApedia enables refined queries to identify protein–RNA complexes, based on various biological, structural, and annotation criteria. The advanced search interface (Figure 3) is designed to offer flexibility in defining queries while maintaining clarity and control over the returned results.
In Figure 3, the user can enter a search term in the main field and start the query using the Search button (1). Searches use partial matching and are case-insensitive, being applied exclusively to the selected field, which ensures greater accuracy in retrieving entries. The interface allows selection of only one search criterion at a time (Figure 3, 3), avoiding ambiguities in interpreting results. Available criteria include RNA sequence, protein sequence, PDB identifier, RNA organism, protein organism, RNA type, RNA modifications, Pfam-annotated RNA-binding domains, and RNA type classification method.
Advanced Search (Figure 3) also allows downloading results in CSV format via the Download CSV button (2). This functionality enables direct reuse of data for statistical analyses, external computational pipelines, and large-scale comparative studies.

After performing a query using Advanced Search, the retrieved protein–RNA complexes are displayed in a dynamic results table (Figure 4), enabling detailed inspection and efficient navigation of the returned data.
As illustrated in Figure 4, the total number of entries found is explicitly displayed (1), allowing the user to assess the scope of the query performed immediately. This information is particularly useful for broad searches, such as those based on general RNA types.
The table interface allows control over the number of entries displayed per page using the Show entries option (2), facilitating navigation in extensive datasets. Furthermore, the table provides column sorting and filtering, allowing the user to refine the results visualization based on specific criteria.
Each row in the results table represents an individual protein–RNA pair and presents essential information, including the RNApedia internal identifier, the PDB ID, the structural resolution, the RNA type, and a brief description of the complex. From this table, the user can access the individual page of each complex through the View option, continuing the structural, sequential, and interaction analysis. In an integrated manner, the results displayed in Figure 4 can be exported in CSV format, enabling reuse in statistical analyses, computational pipelines, and large-scale comparative studies.

2.4 Explore
In addition to advanced search, RNApedia offers a general exploration page (Explore), which allows direct navigation through all protein–RNA complexes available in the database. This interface provides a panoramic view of the data, enabling quick inspection, sorting, and filtering of entries (Figure 5).
As illustrated in Figure 5, the exploration table includes a global text search field (1), which allows dynamically filtering the displayed entries based on keywords present in the table columns. This functionality is useful for quickly inspecting recurring terms, without the need to define specific advanced search criteria.
Each row in the table represents an individual protein–RNA pair, identified by an RpID (RNApedia entry ID) (2). The RpID is the unique identifier for each RNApedia entry and consists of an eight-character code in the format:
PDB_ID–RNA_CHAIN–PROTEIN_CHAIN
For example, the identifier 1ABC_A_B indicates a complex derived from the 1ABC structure of the PDB, where strand A corresponds
to RNA and strand B to protein. This scheme ensures the unambiguous identification of each protein–RNA pair, even when multiple strands are present in the same
structure.
In addition to the RpID, the table presents relevant structural and functional information, including experimental resolution, structure title, RNA size, protein size, RNA type, and the number of protein–RNA contacts identified. The interface also allows controlling the number of entries displayed per page and sorting the results according to different columns, facilitating exploratory data analysis.
By selecting a specific RpID, the user is directed to the individual page of the complex, where structural, sequence, and interaction information is presented in detail. Thus, the Explore page (Figure 5) complements the advanced search functionalities by offering a broad and interactive navigation tool, suitable for initial exploratory analyses and general inspection of the database content.

2.5 Homepage of a complex in RNApedia
The individual page for each complex in RNApedia was designed to integrate, in a single environment, structural visualization, experimental information, and detailed quantitative analyses of the protein–RNA interface. Figure 6 shows this page and its main components, described below.
When accessing an RNApedia entry, the user views a specific protein–RNA pair, identified by an RpID (RNApedia ID). This identifier reflects the database's central strategy, which analyzes interactions at the level of individual protein–RNA pairs.
The RpID is composed of the PDB identifier followed by the identifiers of the RNA and protein chains, in the format:
PDB_ID – RNA_CHAIN – PROTEIN_CHAINExample:
1H4Q_B_T
(1) Three-dimensional visualization of the complex
(Figure 6.1)The panel on the right of the page presents the interactive three-dimensional visualization of the protein–RNA complex. The visualization is fully interactive, allowing rotation, zoom, and detailed inspection of the interaction interface. This visualization serves as a structural reference for all quantitative analyses presented on the page. It can be synchronized with selections made in the sequence and contact panels, facilitating integrated data interpretation.
(2) Protein–RNA contact analysis
(Figure 6.2)The Contacts analysis button provides access to the module for analyzing intermolecular interactions between protein and RNA. In this module, the user can identify hydrogen bonds, hydrophobic, electrostatic, and repulsive interactions; view specific contacts directly in the 3D structure, highlighting the residues and nucleotides involved; and export interaction lists for external analyses.
This feature allows detailed investigation of which protein residues and RNA nucleotides directly participate in the interface, making it particularly useful for functional and comparative studies.
(3) General information about the complex
(Figure 6.3)The central panel of the page presents the structural and experimental metadata of the complex.
Structural identification
- Complex name: functional description of the complex.
- PDB ID: identifier of the structure in the Protein Data Bank.
- Deposition date: date of submission to the PDB.
- Experimental method: technique used for structural determination (e.g., X-ray diffraction or NMR).
- Resolution (Å): resolution of the experimental structure, when applicable.
Composition of the original PDB structure
RNApedia explicitly reports the overall composition of the original PDB file:
- Original PDB: Total chains — total number of molecular chains present in the PDB file.
- Original PDB: Protein chains — number of protein chains in the structure.
- Original PDB: RNA chains — number of RNA chains present.
Protein–RNA interaction metrics
- Total contacts: total number of intermolecular contacts detected between the protein and RNA.
- PDBbind v2020 [3,4] / ProNAB ID [5]: indicates the availability of experimental affinity data from external databases.
When available, the following are displayed:
- Binding affinity (Kd, nM): dissociation constant, where smaller values indicate higher binding affinity.
- Binding free energy (ΔG): binding free energy; negative values indicate energetically favorable interactions.
Accessible and buried surface area
To characterize the protein–RNA interface, RNApedia calculates surface area metrics using the NACCESS software [6,7]:
- Total BSA (Ų): total area buried at the interface after complex formation.
- Polar BSA (Ų): contribution of polar atoms, associated with hydrogen bonds and electrostatic interactions.
- Non-polar BSA (Ų): hydrophobic contribution of the interface.
These values help understand how the interface is stabilized and the relative roles of polar and hydrophobic interactions.
Interface coverage
- Protein interface coverage (%): fraction of the total protein surface involved in the interaction with RNA.
- RNA interface coverage (%): fraction of the RNA surface in contact with the protein.
These metrics allow comparisons of the relative involvement of each molecule in complex formation.
Ligands and ions
- Ligands: small molecules bound to the complex (e.g., ATP).
- Ions: metal ions or structural cofactors present (e.g., Zn²⁺).

Download options on the complex page
RNApedia offers download options for each protein–RNA complex, allowing users to obtain structural files, sequences, and analytical reports for external analyses. Figure 8 illustrates the download menu available on the complex page, with its components described below.
Downloading the PDB file processed by RNApedia
(Figure 7.1)The PDB option allows downloading the PDB file corresponding to the protein–RNA pair defined by RNApedia. This file contains only the RNA and protein chains selected for the RpID. It differs from the original PDB by removing chains that are not relevant to the analyzed pair. This standardization ensures consistency between the structural data and the metrics presented on the page.
Accessible Surface Area (ASA) Report
(Figure 7.2)The ASA report option generates a detailed text file containing the Accessible Surface Area (ASA) calculations performed for the complex.
Download of structural and sequence formats
(Figure 7.3)The lower block of the download menu brings together different structural and sequence file formats:
- mmCIF — complete original structural file in mmCIF format, recommended for large structures and compatible with modern tools.
- Full PDB — complete original PDB file, exactly as deposited in the PDB, including all chains present in the experimental structure.
- FASTA — sequence file containing the protein sequence and the RNA sequence corresponding to the RpID-defined pair.
These options allow easy integration of RNApedia data into external pipelines for analysis, sequence alignment, structural modeling, or machine learning.

Accessible Surface Area (ASA) Report
As described previously, RNApedia provides a detailed Accessible Surface Area (ASA) report for each protein–RNA complex. This report is automatically generated and can be accessed through the ASA report option available in the download menu of each complex page.
Global summary of ASA and buried surface area
(Figure 8.1)The first section of the ASA report presents a global summary of surface area metrics, calculated for the protein–RNA complex and for its individual components.
What is ASA?
Accessible Surface Area (ASA) quantifies the portion of a molecular surface that is exposed to the solvent. In RNApedia, ASA values are calculated separately for:
- the isolated protein,
- the isolated RNA,
- the protein–RNA complex.
Additionally, the report distinguishes between contributions from polar atoms and non-polar (hydrophobic) atoms, allowing a detailed characterization of the interaction interface.
Information presented in this section
- Structure ID: the complex identifier in RNApedia format (RpID).
- Global ASA and Buried Surface Area (all atoms): total ASA of the isolated protein; total ASA of the isolated RNA; ASA of the protein–RNA complex; ΔASA (change upon complex formation); BSA (surface area buried at the interface).
- Non-polar (hydrophobic) ASA and BSA: quantifies hydrophobic contribution to the interface.
- Polar ASA and BSA: describes the contribution of polar atoms (hydrogen bonds and electrostatic interactions).
Together, these measurements provide a quantitative description of the size, composition, and physicochemical nature of the protein–RNA interaction interface.
Per-residue ASA report
(Figure 8.2)The second section of the file contains the per-residue ASA report, which details the individual contribution of each protein residue to the interaction interface. Each row typically includes residue type (RES), chain identifier, residue number, total ASA, polar ASA, and non-polar ASA.
This level of detail allows users to:
- identify residues directly involved in the interface,
- detect interaction hotspots,
- compare interface composition across different complexes,
- support structure-based analyses such as mutagenesis or molecular modeling.

RNA section
(Figure 9)In addition to global complex information, RNApedia provides sections dedicated exclusively to RNA and protein, allowing detailed analysis of the sequential, structural, and functional properties of each molecule individually. These sections integrate data derived from external databases, specialized tools, and standardized computational analyses, maintaining consistency with the protein–RNA pair defined by the RpID.
The RNA section presents information on sequence, composition, classification, modifications, and secondary structure, as well as detailed reports accessible for download.
RNA report
(Figure 9.1)The RNA report option provides a complete report containing detailed analyses of the RNA secondary structure, automatically generated by RNApedia. This report consolidates information from the DSSR [8] and RNAfold tools [9].
RNA identification and classification
(Figure 9.2)- RNA chain ID: identifier of the RNA chain in the complex.
- RNA description: functional description or structural annotation of the RNA.
- Source organism: organism of origin of the molecule.
- RNA type: functional type of RNA (e.g., tRNA, rRNA, mRNA).
- RNA classification: performed using a hierarchical approach: (1) RNAcentral [10–12], (2) Infernal (inference of RNA alignments) [13], and (3) manual curation. The used method is explicitly indicated in the Classification method field.
RNA length, modifications, and composition
(Figure 9.3)- RNA length (nt): total length of the sequence in nucleotides.
- RNA modifications: list of modified nucleotides (e.g., 5MU, PSU).
- GC content (%): percentage of G and C.
- Base composition (A|C|G|U): absolute number of each base.
RNA sequence and secondary structure
(Figure 9.4)- RNA sequence: presented interactively; nucleotides can be selected and highlighted in the 3D structure.
- Dot-bracket (DSSR): experimental secondary structure from the 3D geometry.
- Dot-bracket (RNAfold): predicted secondary structure with free energy (ΔG).
- Free energy (kcal/mol): more negative values indicate higher predicted stability.
RNA report – file details
(Figure 10)The RNA report generated by RNApedia provides an in-depth characterization of the RNA molecule at the level of sequence composition, secondary structure organization, and thermodynamic stability. The file is structured into blocks, each focusing on a specific aspect of RNA architecture.
Structural summary (DSSR)
(Figure 10.1)The first block corresponds to the global structural summary derived from DSSR. It includes total nucleotides, number of base pairs, helices/stems, hairpins, internal loops, junctions, and special interactions such as wobble pairs (e.g., G–U) and stacking.
Nucleotide composition and base pair classification
(Figure 10.2)This block includes absolute nucleotide counts (A, C, G, U) and base-pair classification using the Leontis–Westhof scheme, describing interacting edges (Watson–Crick, Hoogsteen, Sugar) and cis/trans orientation.
Structural elements
(Figure 10.3)Summarizes secondary structure motifs along the sequence, listing structural elements with nucleotide ranges to facilitate mapping between structure and sequence.
Free energy and structural details
(Figure 10.4)Provides thermodynamic and structural refinement with detailed secondary structure elements and free energy values (ΔG), typically derived from RNAfold predictions. Negative ΔG values indicate energetically favorable conformations.

Protein section
(Figure 11)The protein section provides an integrated view of the protein component of each protein–RNA complex, combining sequence information, physicochemical properties, structural annotation, and functional domain mapping. RNApedia also offers a downloadable protein report containing detailed structural analyses.
Protein report
(Figure 11.1)The Protein report option allows users to download a detailed, automatically generated report summarizing the protein’s secondary structure and functional architecture.
Protein identification and physicochemical properties
This panel summarizes fundamental protein-chain characteristics:
- Protein chain ID
- Protein description
- Source organism
- UniProt ID
- Protein length (aa)
- Molecular weight (kDa)
- Isoelectric point (pI)
- Average hydrophobicity
- Aromaticity
- Instability index
Together, these parameters provide a concise physicochemical profile of the protein, facilitating comparisons across different complexes.
Protein sequence and functional motifs
(Figure 11.2)The protein sequence is presented in a fully interactive format, tightly integrated with the 3D visualization of the complex. Residues or regions can be selected and synchronized with the three-dimensional structure. Multi-selection is supported (e.g., Ctrl/Cmd for multi-selection, Shift for intervals).
Functional motifs (Pfam) are identified using the Pfam database and displayed directly on the sequence track (Figure 12.3), enabling direct interpretation of functional domains involved in RNA binding.

Protein report – file details
(Figure 12)Secondary structure per residue
(Figure 12.1)Provides residue-by-residue secondary structure assignment computed using STRIDE [14], including secondary structure type, dihedral angles φ and ψ, chain identifier, and residue position.
Global secondary structure statistics
(Figure 12.2)Includes a quantitative summary of the protein’s overall structural composition, such as percentage and absolute counts of α-helices, β-strands, and coil/loop regions.
Structural motifs and domains (Pfam)
(Figure 12.3)Lists all functional domains identified via Pfam, including domain name/description, accession number, residue boundaries, and confidence scores (e.g., E-values), enabling correlation between protein architecture and RNA binding.

Contact Analysis – Protein–RNA interactions
The Contact Analysis page presents a detailed analysis of molecular interactions between protein and RNA for each protein–RNA pair defined in RNApedia. Interactions are calculated at the atomic level and integrated into an interactive 3D visualization, allowing simultaneous exploration of tabular and structural data.
Description of interface elements
(Figure 13.)Interaction list (Figure 13.1)
This panel presents the complete list of detected interactions between the protein and RNA. Each row includes:
- Protein chain (Chain1)
- Protein residue (Res1) and atom (Atom1)
- RNA chain (Chain2)
- RNA nucleotide (Res2) and atom (Atom2)
- Interatomic distance (Å)
- Interaction type
Interactions can be filtered by category, including:
- HB — Hydrogen bonds
- HY — Hydrophobic contacts
- SB — Salt bridges
- AT — Attractive interactions
- RE — Repulsive interactions
- DS — Disulfide bonds
- AS — Aromatic stacking
- WA — Water-mediated contacts
- IO — Ion-mediated contacts
- OT — Other interactions
The list can be exported in CSV format, enabling external quantitative analysis and reproducibility.
3D visualization of interactions (Figure 13.2)
Displays the interactive three-dimensional visualization synchronized with the interaction list. It supports different representation modes (cartoon, stick, sphere), color/opacity adjustment, molecular surface display, and automatic highlighting of residues/atoms when selecting an interaction.
Atomic-level interaction detail (Figure 13.3)
Shows a zoomed inspection of the selected interaction, including precise interatomic distance and interaction type. This is useful for geometric validation and mechanistic studies.
Contact map (Figure 13.4)
The Contact map tool generates a matrix representation of interactions between protein residues and RNA nucleotides, providing a global view of the interface and enabling pattern analyses and comparisons across complexes.
Download interactions (Figure 13.5)
Allows downloading the complete interaction dataset identified for the complex, ensuring direct access to the raw data used in visualization and analysis.

3. Contact Analysis
Interatomic contacts represent fundamental biochemical interactions that occur between macromolecules and play a central role in stabilizing molecular structures and complexes. In protein–RNA systems, these interactions are particularly important for defining binding specificity, molecular recognition, and functional regulation. Computational identification of contacts allows systematic characterization of these interactions at atomic resolution.
In RNApedia, contacts are defined based on distance-based criteria, the most widely used approach for contact detection, in which two atoms are considered interacting when the distance between them is below a predefined cutoff value [15]. Although individual interactions such as hydrogen bonds or hydrophobic contacts are relatively weak, their cumulative effect is essential for the structural stability of protein–RNA complexes [16,17].
Computational framework for contact detection
To ensure a consistent and chemically accurate description of protein–RNA interactions, RNApedia integrates several computational tools and libraries:
RNA definition and modifications
RNA molecules, including all mapped nucleotide modifications, are explicitly defined using the RDKit library [19], which provides a robust cheminformatics framework for molecular representation.
Atom-type classification
For the four standard ribonucleotides (A, C, G, U), atom types are assigned using the LUNA library [18], which enables a detailed characterization of atomic physicochemical properties relevant to molecular interactions. Atoms are classified according to:
- Hydrogen bond donors
- Hydrogen bond acceptors
- Hydrophobic atoms
- Aromatic atoms
- Positively charged atoms
- Negatively charged atoms
The same classification scheme is applied to modified nucleotides, whose atom properties are computed using RDKit [19] to ensure compatibility with the standard nucleotide definitions.
Contact calculation engine
The COCαDA scripts [20,21] were adapted and extended to incorporate the atom-type classification of nucleotides and their modifications. These modified scripts are then used to calculate interatomic contacts between protein and RNA chains.
Protein–RNA pairs that do not present any detectable contacts are excluded from the database. After this filtering step, the final RNApedia dataset comprises 56,133 protein–RNA interaction entries, each supported by explicit atomic contact information.
3.1 Contact Analysis interface
The contact analysis tool can be accessed directly from an individual RNApedia entry for already calculated RNApedia entries, where the user can submit the complexes they are interested in analyzing, or through the independent Atomic Contact Analysis page. The display layout and interaction logic are consistent with those used throughout the platform, ensuring a uniform user experience.
Entry page for contact analysis
(Figure 14)Users can start a contact analysis using one of two input options:
-
Upload a structure file
(Figure 14.1) A local structure file in PDB or CIF format can be uploaded directly through the interface. -
Enter a PDB ID
(Figure 14.2) Alternatively, users can provide a valid PDB identifier, which will be automatically retrieved and processed by RNApedia.
After selecting one of these options, the contact calculation is initiated by clicking on Calculate Contacts.

3.2 Contact analysis results page
(Figure 15)Once the calculation is complete, RNApedia presents the results in an interactive visualization environment composed of several coordinated panels.
Interaction list
(Figure 15.1)This panel displays a comprehensive table of all detected protein–RNA contacts. For each interaction, the following information is provided:
- Protein chain and residue
- Protein atom
- RNA chain and residue
- RNA atom
- Interatomic distance (Å)
- Interaction type
Contacts are classified into categories such as:
- Hydrogen bonds (HB)
- Hydrophobic interactions (HY)
- Salt bridges (SB)
- Attractive and repulsive interactions
Interactive filters allow users to isolate specific interaction types, and the table can be exported as a CSV file for downstream analysis.
3D visualization
(Figure 15.2)The central panel shows an interactive three-dimensional representation of the protein–RNA complex. Key features include:
- Multiple rendering styles (cartoon, stick, sphere)
- Color schemes and opacity controls
- Optional surface representation
- Direct synchronization with the interaction list
Selecting an interaction in the table automatically highlights the corresponding atoms in the 3D structure, enabling intuitive exploration of contact geometry. This view provides a clear structural interpretation of individual interactions at atomic resolution.
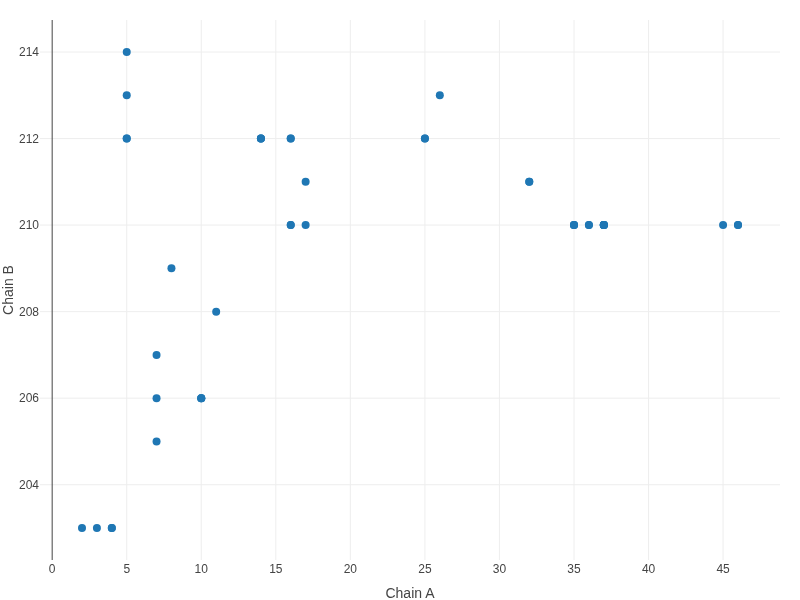
Contact map
(Figure 15.3, Figure 16)The contact map offers a two-dimensional overview of interactions between protein and RNA residues. Each point represents a detected contact, plotted according to the residue indices of the interacting chains. This representation facilitates the identification of interaction hotspots and global binding patterns.
Download options
(Figure 15.4)Users may download:
- The full list of detected interactions (CSV)
- The processed PDB structure used for contact analysis

Conclusion
RNApedia is presented as a comprehensive and integrated platform for the structural analysis of protein–RNA complexes, combining curated structural data with robust computational tools and interactive visualizations. By integrating sequence, structural, physicochemical, and molecular interaction information into a single environment, RNApedia enables both global overviews and detailed atomic-level investigations of protein–RNA interfaces.
The availability of dedicated RNA and protein reports, together with consolidated metrics such as accessible surface area (ASA), secondary structure assignment, functional motifs, and detailed contact analyses, ensures methodological consistency and comparability across complexes. The RNA classification strategy combining RNAcentral annotations, Infernal-based inference, and manual curation provides broad coverage and reliability, particularly for complexes containing modified or poorly characterized RNAs. Complementarily, protein functional motifs annotated using the Pfam database allow direct association between protein functional regions and their involvement in RNA recognition and binding.
The contact analysis tool represents a key contribution of RNApedia, employing a refined methodology for identifying and classifying interatomic and inter-residue interactions based on physicochemical properties of both standard and modified nucleotides. This approach enables a more realistic description of the forces stabilizing protein–RNA complexes, going beyond purely geometric distance-based criteria. The combined use of interaction tables, three-dimensional visualizations, and contact maps provides multiple complementary perspectives, facilitating biological interpretation and downstream computational applications, including structural modeling and machine learning.
Overall, RNApedia establishes itself as a valuable resource for the structural biology and bioinformatics communities, supporting advances in the understanding of protein–RNA interactions and providing a high-quality, large-scale dataset suitable for functional, comparative, and data-driven studies.
References
- Hamelryck, T. & Manderick, B. PDB file parser and structure class implemented in Python. Bioinformatics 19, 2308–2310 (2003).
- Fierro-Monti, I. RBPs: an RNA editor’s choice. Front. Mol. Biosci. 11 (2024).
- Wang, R., Fang, X., Lu, Y. & Wang, S. The PDBbind Database: Collection of Binding Affinities for Protein–Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 47, 2977–2980 (2004).
- Wang, R., Fang, X., Lu, Y., Yang, C.-Y. & Wang, S. The PDBbind Database: Methodologies and Updates. J. Med. Chem. 48, 4111–4119 (2005).
- Harini, K., Srivastava, A., Kulandaisamy, A. & Gromiha, M. M. ProNAB: database for binding affinities of protein–nucleic acid complexes and their mutants. Nucleic Acids Res. 50, D1528–D1534 (2022).
- Hubbard, S. J. & Thornton, J. M. NACCESS computer program. Department of Biochemistry and Molecular Biology, University College London (1993).
- Ding, J. & Arnold, E. NACCESS. In: International Tables for Crystallography, 685–694 (2006).
- Lu, X.-J., Bussemaker, H. J. & Olson, W. K. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 43, e142 (2015).
- Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
- The RNAcentral Consortium. RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 47, D221–D229 (2019).
- The RNAcentral Consortium. RNAcentral: an international database of ncRNA sequences. Nucleic Acids Res. 43, D123–D129 (2015).
- RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 45, D128–D134 (2017).
- Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
- Heinig, M. & Frishman, D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32, W500–W502 (2004).
- Silveira, C. H. da. Protein cutoff scanning: aplicação da varredura exaustiva de distâncias inter-resíduos na análise de contatos intracadeia em proteínas globulares. (2008).
- Silva, M. F. M. et al. Proteingo: Motivation, user experience, and learning of molecular interactions in biological complexes. Entertain. Comput. 29, 31–42 (2019).
- Fassio, A. V., Santos, L. H., Silveira, S. A., Ferreira, R. S. & de Melo-Minardi, R. C. nAPOLI: A Graph-Based Strategy to Detect and Visualize Conserved Protein-Ligand Interactions in Large-Scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1317–1328 (2020).
- Fassio, A. V. et al. Prioritizing Virtual Screening with Interpretable Interaction Fingerprints. J. Chem. Inf. Model. 62, 4300–4318 (2022).
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling.
- Lemos, R. P., Mariano, D., Silveira, S. D. A. & de Melo-Minardi, R. C. COCαDA - a fast and scalable algorithm for interatomic contact detection in proteins using Cα distance matrices. Front. Bioinforma. 5 (2025).
- Lemos, R. P., Mariano, D., Silveira, S. A. & de Melo-Minardi, R. C. COCαDA - Large-Scale Protein Interatomic Contact Cutoff Optimization by Cα Distance Matrices. In: Simpósio Brasileiro de Bioinformática (BSB), 59–70 (SBC, 2024). doi:10.5753/bsb.2024.245545.
How to cite
If you use RNApedia, please cite the following reference:
BASTOS, Luana Luiza et al.
A strategy for refining the calculation of contacts in protein–RNA complexes.
In: Brazilian Symposium on Bioinformatics (BSB). SBC, 2024. pp. 241–246.
Additional citations
If RNApedia is used, please also cite the following references, as appropriate:
- Dunin-Horkawicz, S. et al. MODOMICS: a database of RNA modification pathways. Nucleic Acids Research, 34(suppl_1), D145–D149 (2006).
- Boccaletto, P. et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Research, 50(D1), D231–D235 (2022).
- Nawrocki, E. P.; Kolbe, D. L.; Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics, 25(10), 1335–1337 (2009).
- Hubbard, S. J.; Thornton, J. M. NACCESS: a program for calculating accessibilities. University College London (1992).
- RNAcentral Consortium. RNAcentral: an international database of ncRNA sequences. Nucleic Acids Research, 43(D1), D123–D129 (2015).
- RNAcentral Consortium. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Research, 45(D1), D128–D134 (2017).
- RNAcentral Consortium. RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Research, 47(D1), D221–D229 (2019).
- Lu, X.-J.; Bussemaker, H. J.; Olson, W. K. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Research, 43(21), e142 (2015).
- Gruber, A. R. et al. The Vienna RNA websuite. Nucleic Acids Research, 36(suppl_2), W70–W74 (2008).

