
Documentation
What is COCαDA-web?
COCαDA-web is a user-friendly web interface for using the COCαDA command line tool. COCαDA-web also contains a database of pre-calculated contacts for all structures available in the PDB (Protein Data Bank), which is updated weekly.
What is COCαDA?
COCαDA (COntact search pruning by Cα Distance Analysis) accelerates the calculation of atomic interactions in proteins, by using a set of fine-tuned Cα distances between every pair of aminoacid residues. The tool includes a customized parser for both PDB and CIF files, with support for large files, residue and atom filtering, and geometric analysis (e.g., centroids and normal vectors for aromatic residues). Users can also define their own contact distance cutoffs, and define pH-specific values for electrostatic interactions.
The contact types available are:
- Hydrophobic
- Hydrogen Bond
- Attractive
- Repulsive
- Disulfide Bond
- Salt Bridge
- Aromatic Stacking
Contact rules
| Contact Type | Distance range (Å) | Description | Acronym |
|---|---|---|---|
| Hydrogen Bond | 0 ≤ dist ≤ 3.9 | Acceptor and Donor atom pair | HB |
| Disulfide Bond | 0 ≤ dist ≤ 2.8 | Cys:SG atom pair | DS |
| Hydrophobic | 2.0 ≤ dist ≤ 4.5 | Hydrophobic atom pair | HY |
| Repulsive | 2.0 ≤ dist ≤ 6.0 | Equally charged atoms | RE |
| Attractive | 3.9 ≤ dist ≤ 6.0 | Differently charged atoms | AT |
| Salt Bridge | 0 ≤ dist ≤ 3.9 | Equally charged atoms AND hydrogen bonding | SB |
| Aromatic Stacking | 2.0 ≤ dist ≤ 5.0 | Centroids of two aromatic rings in parallel or perpendicular orientation |
AS |
| Uncertain Electrostatic | Same as AT, RE, SB | Check the description here | uAT, uRE, uSB |
How to use COCαDA-web
Home Page
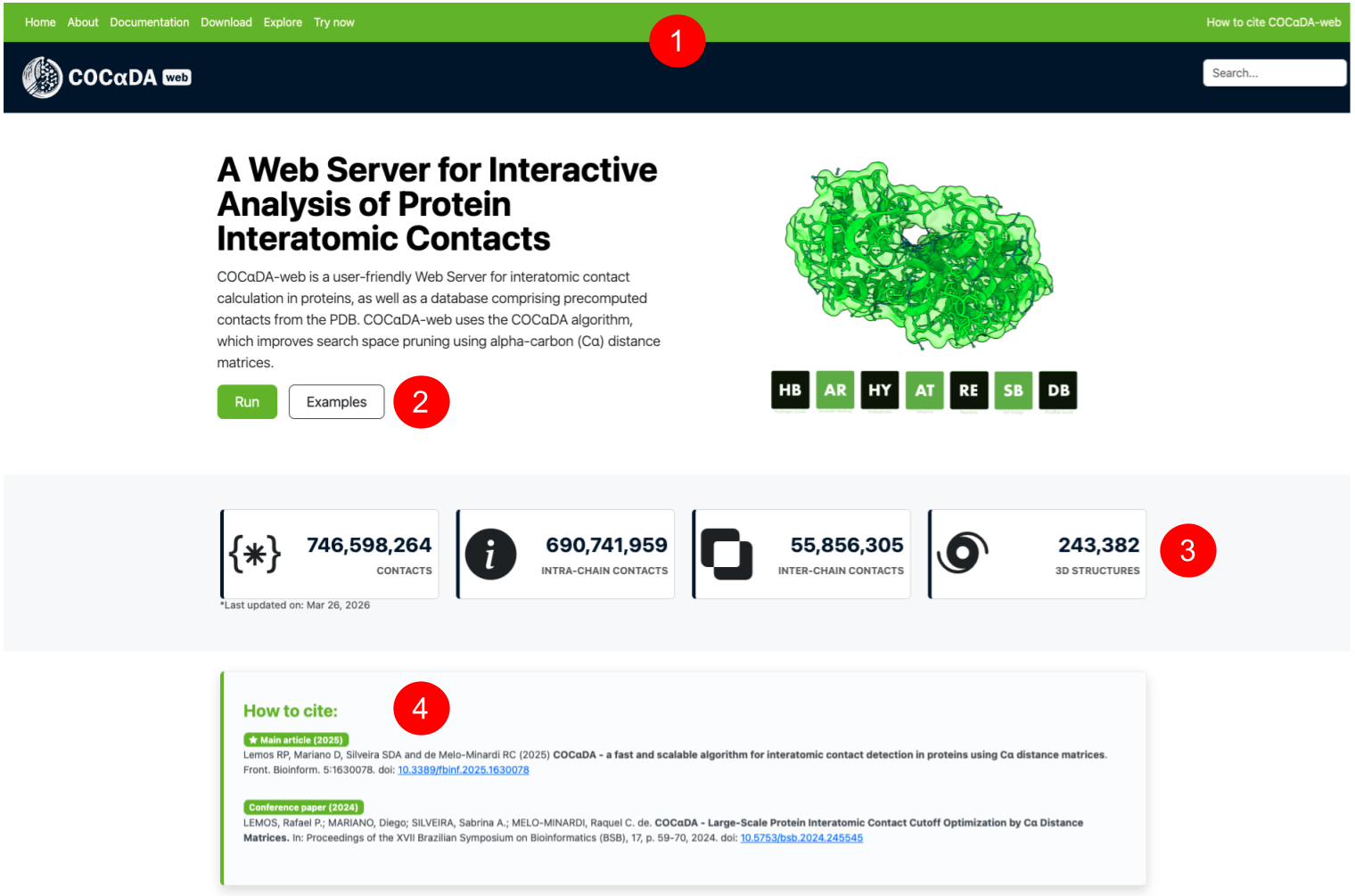
(1) Navigation and search bars. (2) Short description of the tool, with buttons to run it or see examples. (3) Statistics for the precomputed contacts from the PDB, with total, inter-, and inter-chain numbers, and the number of current processed structures (up to 10,000 residues). The statistics are updated each week automatically. (4) Citation guidelines for the COCαDA tool.
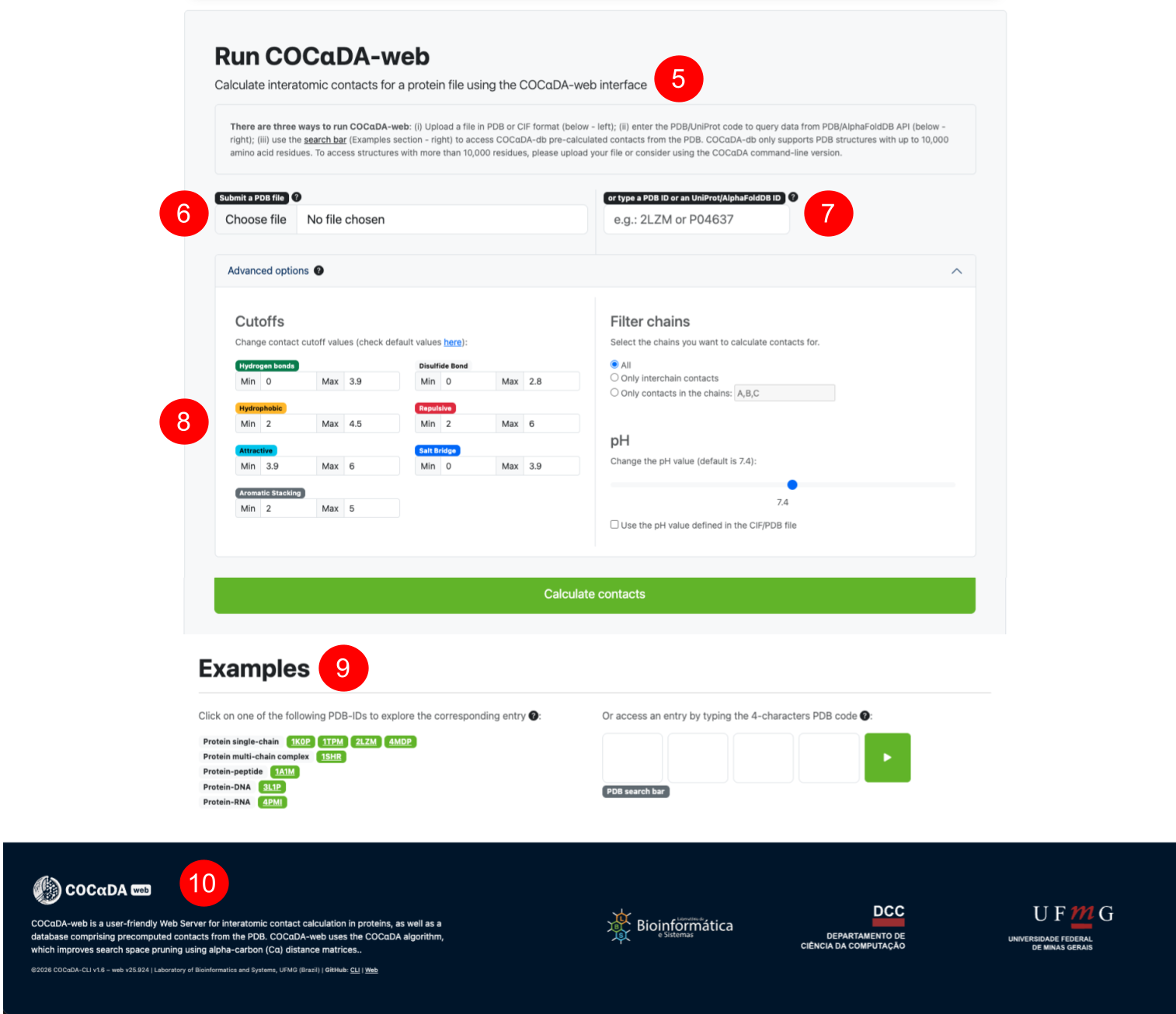
(5) Instructions on the three different ways to use COCαDA-web, which are: user-submitted PDB/mmCIF file (6); PDB/Uniprot ID (7); searching for pre-computed proteins from the PDB (1). (8) Advanced options for COCαDA-web, including user-specified cutoff values for each different type of contact; filtering by specific chains or only inter-chain contacts; and defining the pH value to be used for electrostatic contacts. (9) Examples section, containing different sets of proteins from the PDB. (10) Short description of the Web Server and current version for COCαDA and COCαDA-web.
Explore Page
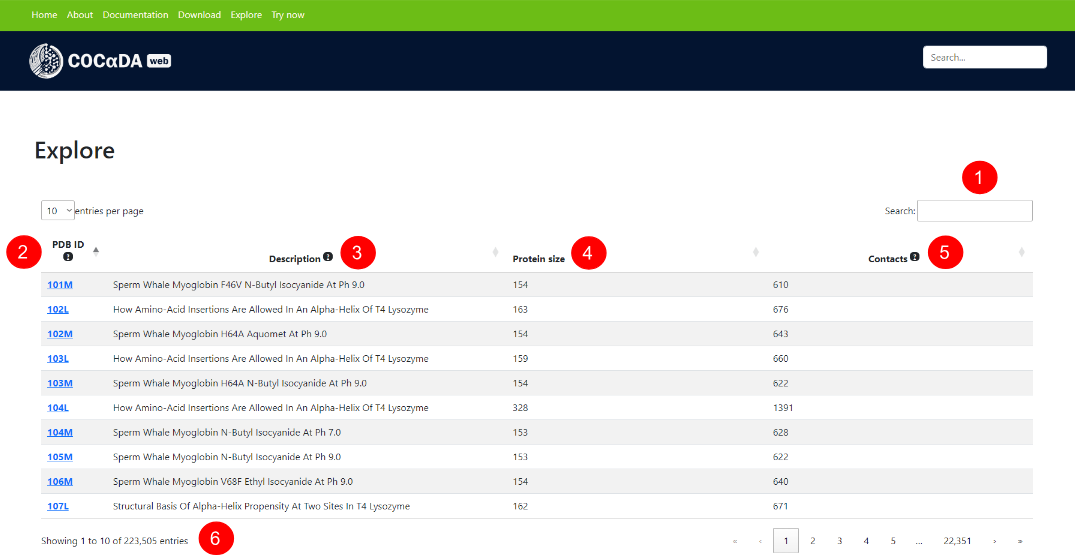
The explore page contains all proteins in the PDB up to 10,000 residues. (1) Search and filtering bar, which works for all the columns in the table. (2) PDB ID column. (3) Description column, containing the protein name derived from the PDB. (4) Protein size, number of contacts, and pH. For the pH column, the value shown was obtained directly from the PDB, when available, defaulting to 7.4 when not defined in the deposition. (5) Number of proteins in the current filter and pagination of the results.
Results Page
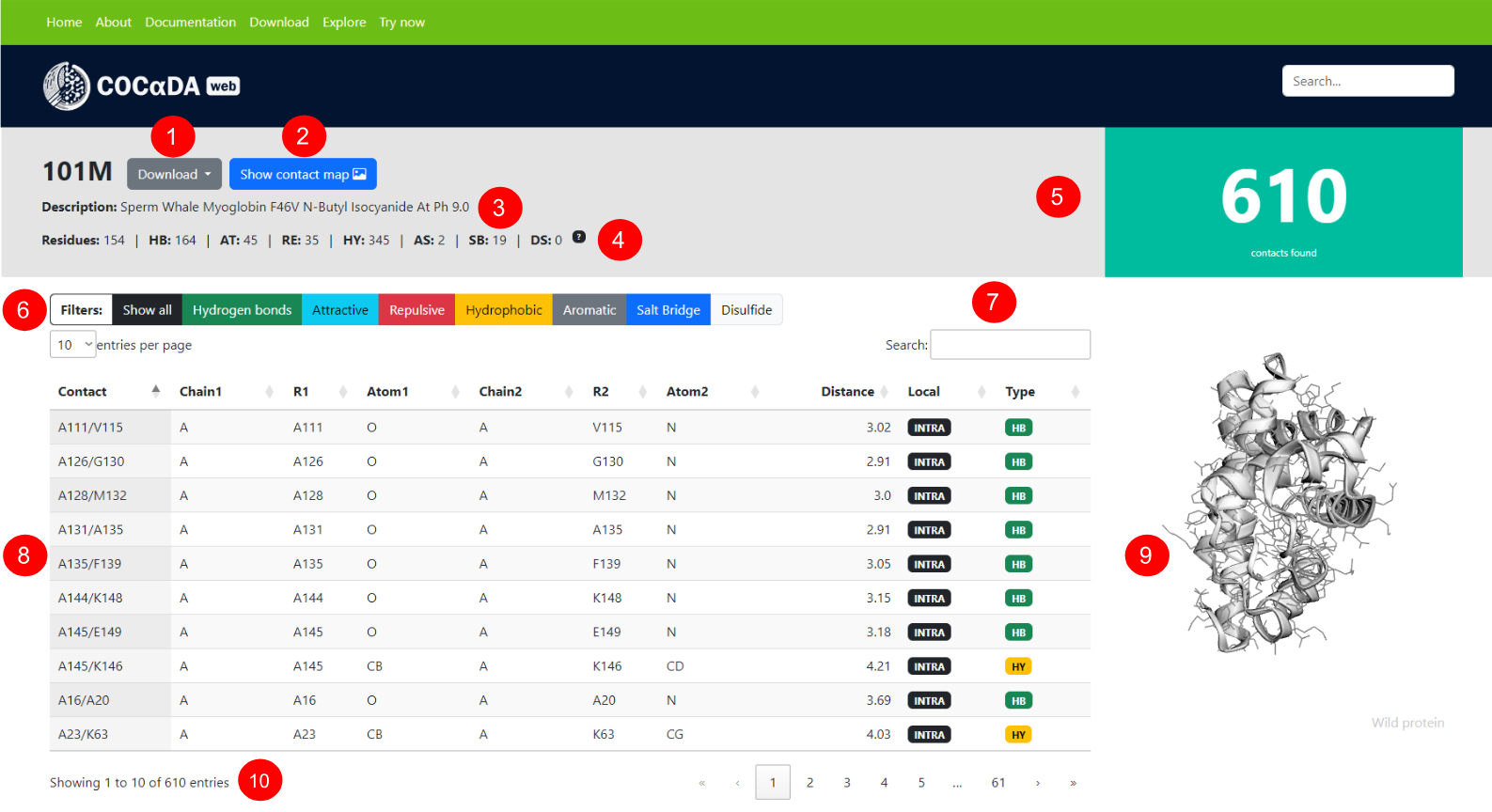
(1) Download options for the results, which include the contact list in CSV format, the submitted PDB/mmCIF file, and the Pymol session with annotated contacts in PSE format. (2) Button to show the protein contact map in a pop-up window. (3) Protein description, obtained from the PDB. (4) Full information for the protein, including the number of residues, number of contacts of each individual type, and pH used for calculation.(5) Total number of contacts identified for the protein. (6) Options for filtering the results. (7) Full list of contacts, with the columns being: contact name; protein chain of the first atom; residue of the first atom; name of the first atom; protein chain of the second atom; residue of the second atom; name of the second atom; distance between the atom pair, in angstroms; localization of the contact (intra-chain or inter-chain); contact type; show the individual contact in the visualization.(8) Interactive visualization of the protein. (9) Number of contacts in the current filter and pagination of the results.
HY = hydrophobic; HB = hydrogen bond; AT = attractive; RE = repulsive; SB = salt bridge; AS = aromatic stacking; uAT = uncertain attractive; uRE = uncertain repulsive; uSB = uncertain salt bridge.
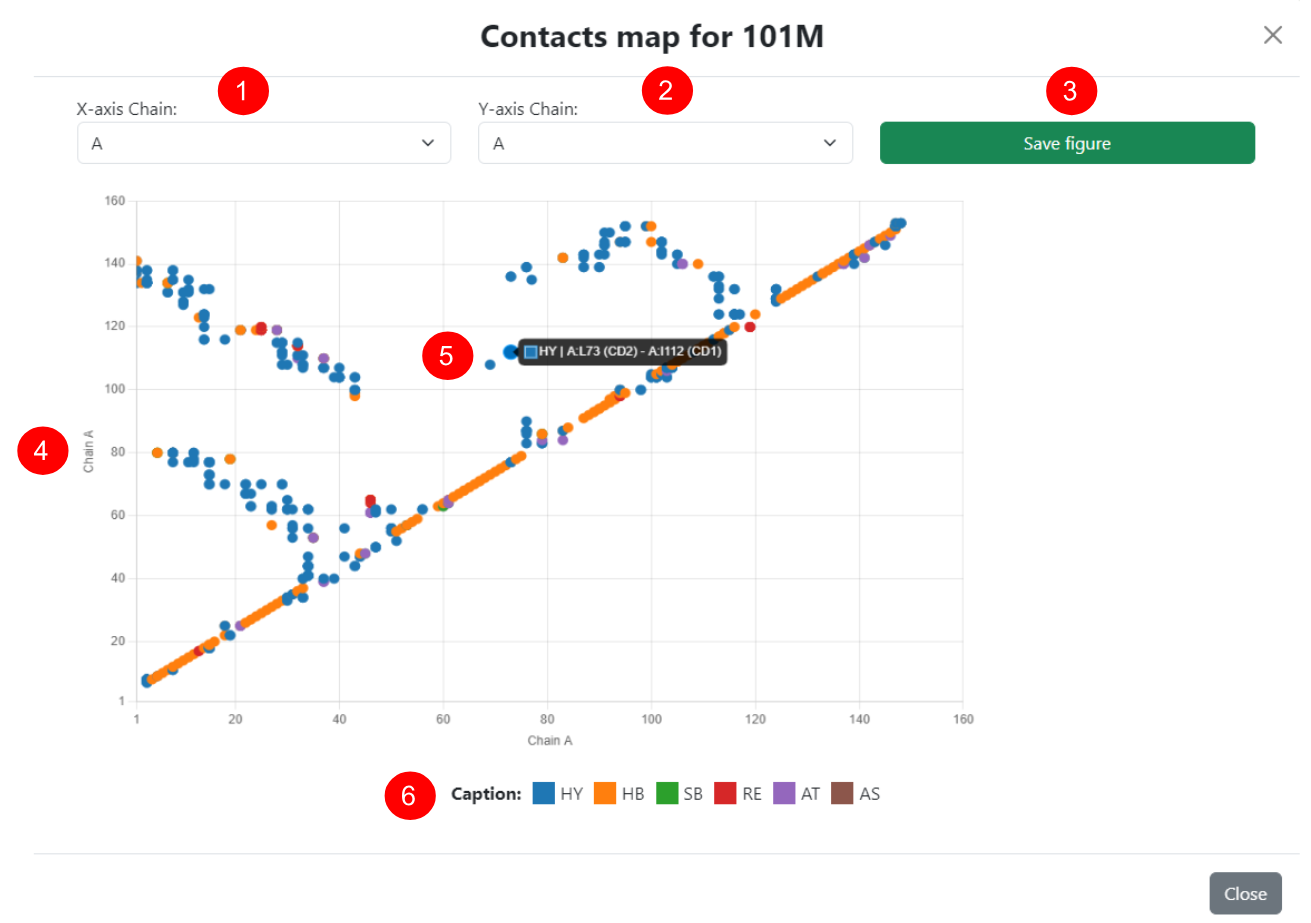
Contact Map
Next to the download button on the results page, the user can view the protein contact map in an interactive pop-up window. Since this is a two-dimensional representation of a protein's contacts, each axis of the map represents a polypeptide chain, both of which can be dynamically adjusted (1 for the X-axis and 2 for the Y-axis). In this way, inter-chain contacts can be visualized by selecting different chains in the menus, and the maps can also be saved in ".png" format (3). (4) Interactive view of the selected pair of chains, with each point being a pair of residues, and the colors representing the predominant type of contact for the pair. (5) By hovering the mouse over any of the points, the user can see the detailed information for all the contacts made by the residue pair.
(6) Color scheme for the contact types. HY = hydrophobic; HB = hydrogen bond; AT = attractive; RE = repulsive; SB = salt bridge; AS = aromatic stacking; uAT = uncertain attractive; uRE = uncertain repulsive; uSB = uncertain salt bridge.
Types of Bonds and Interactions used in COCαDA-web
Disulfide Bonds
Disulfide bonds are one type of covalent bonds present in proteins, yet they are still weaker than peptide bonds. Formed exclusively by the sulfur atoms of the thiol (-SH) groups from a pair of cysteine residues, disulfide bonds are extremely important in the folding process and stability of certain proteins (Sevier2002).
Since the intracellular environments of living organisms are predominantly reducing, proteins containing disulfide bridges tend to be unstable in the cellular cytosol. As a result, the formation of these bonds typically occurs in specific regions and in the presence of catalysts, such as the endoplasmic reticulum in eukaryotes, the periplasm in prokaryotes, and the intermembrane space in mitochondria (Sevier2002, Hatahet2010).

Hydrogen Bonds
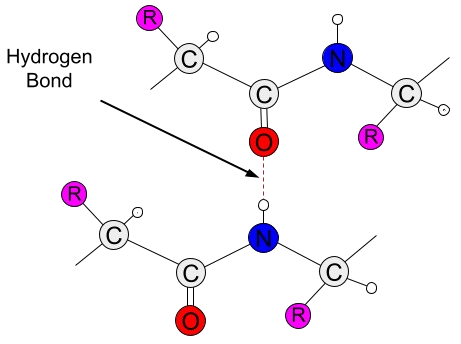
Hydrogen bonds are a type of weak, short-range interaction that can occur between atoms of amino acid residues, playing an essential role in the folding process and functionality of proteins (Saenger1994, Agostini2019). With an electrostatic component, hydrogen bonds arise due to the difference in electronegativity between hydrogen atoms and other more electronegative atoms. In the case of amino acids, the only two atoms electronegative enough are oxygen (O) and nitrogen (N), both of which are key components of amino acids (Nelson2012).
When covalently bonded to one of these atoms, the hydrogen atom's electron cloud shifts toward the bond, creating two poles of opposite charges between them. Due to the partial charge generated on the hydrogen atom by this shift, it can then interact with another electronegative atom that has a partial negative charge. This third atom is called a hydrogen acceptor and forms a weak, attractive bond with the hydrogen atom (Nelson2012, Kessel2018). In addition to occurring between amino acid atoms themselves, hydrogen bonds can also be mediated by water molecules, which make up nearly the entire volume of intra- and extracellular environments in vivo (Saenger1994).
Hydrophobic Interactions
Due to their side chains (-R), amino acids can exhibit polarity properties, making them either polar or nonpolar. In a protein, the combination of these characteristics generates hydrophobic (nonpolar) and hydrophilic (polar) regions within its structure (Camilloni2016). A chemical environment composed solely of water molecules features numerous hydrogen bonds between them, forming a stable structure (Levy2006). However, when any solute (such as a protein) is introduced into this environment, the resulting disturbance breaks hydrogen bonds among nearby water molecules, which then reestablish bonds directly with the solute (Dunn2010, Kessel2018).
Since these interactions can only occur between polar molecules, the nonpolar regions of a protein tend to aggregate within its structure to interact with each other, thereby avoiding contact with water molecules. These interactions between nonpolar molecules are known as hydrophobic interactions and are critically important for protein folding (Kauzmann1959, Pace2011).
Electrostatic Interactions and Salt Bridges
Just like polar and nonpolar side chains, amino acids can also have charged chemical groups in their side chains. This is the case for lysine (K), arginine (R), and histidine (H), which carry positive charges, as well as aspartic acid (D) and glutamic acid (E), which also carry positive charges. Thus, amino acids with the same charge on their side chains form a repulsive electrostatic interaction, whereas those with opposite charges form an attractive electrostatic interaction (Nelson2012).
The charges on the side chains of these five ionizable amino acids play various structural and functional roles, such as pH-mediated protein denaturation, ion transport across membranes, and metal binding (Zhou2018). Additionally, other neutral amino acids can be ionized through the addition of charged chemical groups, as seen in the phosphorylation and dephosphorylation of serine (S), threonine (T), and tyrosine (Y) residues (Hunter2012).
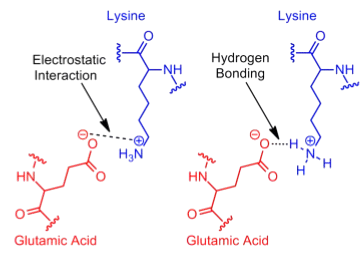
Since electrostatic interactions are a broad category that includes even hydrogen bonds, the term "salt bridge" is commonly used to describe a specific type of attractive electrostatic interaction (Kumar1999, Sinha2002). In salt bridges, the interaction occurs exclusively between fully ionized side chain groups and is further defined as an attractive electrostatic interaction in which at least one of the heavy atoms is within hydrogen bond distance (Donald2011).
Aromatic Stackings
Aromatic stacking occurs exclusively between molecules that contain aromatic groups, also known as aromatic rings. In proteins, these groups are present in three amino acids: phenylalanine (F), tyrosine (Y), and tryptophan (W). The aromatic rings of these amino acids are composed of conjugated double bond systems, in which the electrons of the π orbital are delocalized, providing resonance and stability to the system (Kessel2018).
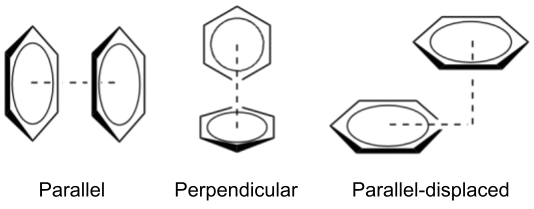
When two aromatic rings interact directly, this is known as π–π stacking, which can be further classified based on the geometry of the interaction (Smith2007). The simplest form is called "face-to-face" (or parallel), where the rings align in a parallel fashion. However, due to the repulsive nature of π-orbital overlap, the parallel geometry is relatively rare (McGaughey1998). The most common interaction patterns are "perpendicular" (T-shaped) and "parallel-displaced", both of which have attractive character (Martinez2012).
Uncertain Electrostatic Contacts
The pH customization feature allows for a more nuanced analysis of electrostatic interactions. We defined pH-sensitive atoms as those located in the side chains of ionizable residues, which can exist in either protonated or deprotonated states (Nelson2012). The protonation state of each sensitive atom is assessed by comparing the user-specified pH value to the residue's characteristic side-chain pKa. To account for the equilibrium between ionic forms, protonation states were classified as ambiguous if |pH - pKa| ≤ 2.0. This threshold, derived from the Henderson-Hasselbalch equation (Henderson1908), ensures that residues with a non-negligible (at least 1%) population of both protonated and deprotonated species are identified (Olsson2011).
This ambiguity directly impacts the assignment of electrostatic contacts. When a pH-sensitive atom is in an ambiguous protonation state, any electrostatic contact it forms, namely attractive, repulsive, or salt bridges, is classified as "uncertain" (designated uAT, uRE, and uSB, respectively). In addition to these charge-based interactions, the thiol group of cysteine residues (C:SG) is treated with special consideration, as its capacity to act as either a hydrogen-bond donor or acceptor is also considered pH-dependent (Marino2010).
Ionizable residues with their corresponding side-chain pKa values and pH-sensitive atoms
| Residue | Side-chain pKa | pH-sensitive atoms |
|---|---|---|
| Aspartic Acid | ~3.9 | OD1, OD2 |
| Glutamic Acid | ~4.3 | OE1, OE2 |
| Histidine | ~6.0 | ND1, NE2 |
| Cysteine | ~8.3 | SG |
| Tyrosine | ~10.1 | OH |
| Lysine | ~10.5 | NZ |
| Arginine | ~12.5 | NE, NH1, NH2, CZ |
References
Agostini, A., Meneghin, E., Gewehr, L., Pedron, D., Palm, D. M., Carbo- nera, D., Paulsen, H., Jaenicke, E., and Collini, E. (2019). How water-mediated hydrogen bonds affect chlorophyll a/b selectivity in Water-Soluble chlorophyll protein. Sci. Rep., 9(1):18255.
Camilloni, C., Bonetti, D., Morrone, A., Giri, R., Dobson, C. M., Brunori, M., Gianni, S., e Vendruscolo, M. (2016). Towards a structural biology of the hydrophobic effect in protein folding. Sci. Rep., 6(1).
Donald, J. E., Kulp, D. W., e DeGrado, W. F. (2011). Salt bridges: geometrically specific, designable interactions. Proteins, 79(3):898–915.
Dunn, M. F. (2010). Protein-Ligand Interactions: General Description. John Wiley & Sons, Ltd, Chichester, UK.
Hatahet, F., Nguyen, V. D., Salo, K. E. H., and Ruddock, L. W. (2010). Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of e. coli. Microb. Cell Fact., 9(1):67.
Henderson, L. J. (1908). Concerning the relationship between the strength of acids and their capacity to displace other acids in an aqueous solution. Am. J. Physiol., 21(1):173–179.
Hunter, T. (2012). Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci., 367(1602):2513–2516.
Kauzmann, W. (1959). Some factors in the interpretation of protein denaturation. In Advances in Protein Chemistry, Advances in protein chemistry, pages 1–63. Elsevier.
Kessel, A. and Ben-Tal, N. (2018). Introduction to proteins: Structure, function, and motion. Chapman & Hall/CRC, Philadelphia, PA.
Kumar, S. e Nussinov, R. (1999). Salt bridge stability in monomeric proteins. J. Mol. Biol., 293(5):1241–1255.
Levy, Y. and Onuchic, J. N. (2006). Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct., 35(1):389–415.
Martinez, C. R. and Iverson, B. L. (2012). Rethinking the term “pi-stacking”. Chem. Sci., 3(7):2191.
McGaughey, G. B., Gagn´e, M., and Rapp´e, A. K. (1998). Π-stacking interactions. J. Biol. Chem., 273(25):15458–15463
Marino, S. M. and Gladyshev, V. N. (2010). Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol., 404(5):902–916.
Nelson, D. L. and Cox, M. M. (2012). Lehninger principles of biochemistry. W.H. Freeman, New York, NY, 6 edition.
Olson, M. H., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput., 7(2):525–537.
Pace, C. N., Fu, H., Fryar, K. L., Landua, J., Trevino, S. R., Shirley, B. A., Hendricks, M. M., Iimura, S., Gajiwala, K., Scholtz, J. M., and Grimsley, G. R. (2011). Contribution of hydrophobic interactions to protein stability. J. Mol. Biol., 408(3):514–528.
Saenger, W. and Jeffrey, G. A. (1994). Hydrogen bonding in biological structures. Springer, Berlin, Germany.
Sevier, C. S. and Kaiser, C. A. (2002). Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol., 3(11):836–847.
Sinha, N. and Smith-Gill, S. (2002). Electrostatics in protein binding and function. Curr. Protein Pept. Sci., 3(6):601–614.
Smith, M. B. e March, J. (2007). March’s advanced organic chemistry. John Wiley & Sons, 6 edition.
Zhou, H.-X. and Pang, X. (2018). Electrostatic interactions in protein structure, folding, binding, and condensation. Chem. Rev., 118(4):1691–1741.

